

How to generate diverse, representative libraries from even the most complex samples
Complex samples can complicate NGS library preparation
Next-generation sequencing has become an essential tool for detecting genetic variations, like SNVs, copy number variants, structural rearrangements, and RNA isoforms, across biological systems. But before any sample reaches the sequencer, it must first undergo NGS library preparation — the process of converting DNA or RNA into sequencer-ready constructs. This step is foundational, and for researchers working with complex or challenging samples, it is often the most critical determinant of downstream data quality.1
Selecting a cost-effective, efficient library preparation chemistry and workflow that is compatible with your sample type can be difficult. Degraded DNA, low-input extracts, non-model organism genomes, and RNA samples can be especially challenging, increasing the likelihood for repeat analyses which can drive up costs and timelines.
In this blog, you’ll learn the basic workflow for NGS library preparation and the potential risks at each step. You will also discover solutions designed to help you generate high-quality, high-coverage libraries from degraded or low-input DNA, avoid GC amplification bias, and improve library diversity. Read on for ways to improve sequencing data quality from even the most complex sample types.
Understanding the NGS library preparation workflow
A standard sequencing library prep workflow involves several interconnected steps, each of which can introduce bias or data loss if not carefully optimized.2 Described below is a generalized library prep workflow; specific platforms or applications may require additional cleanup steps or protocol modifications.
1. Extraction and quality assessment
The library preparation process begins with careful extraction of DNA or RNA from a sample. The choice of method is very important as complex sample types have unique requirements: ancient DNA requires specialized low-pH buffers and rigorous contamination controls; environmental metagenomics samples demand inhibitor removal; FFPE-derived DNA is heavily fragmented and chemically modified; and cell-free DNA (cfDNA) exists at ultra-low concentrations. RNA must be converted to cDNA via reverse transcription, but depending on the specific protocol it can be done before or after fragmentation. Regardless of sample type, accurate assessment of integrity, concentration, and purity is essential before advancing to the next step.
2. Fragmentation
Double-stranded DNA is fragmented to uniform sizes — typically 100-1500 bp, or as determined by the NGS platform — using either enzymatic methods or mechanical shearing. Enzymatic digestion offers more controlled, reproducible fragmentation compared to mechanical shearing, particularly for low-input or degraded samples, and contributes to improved downstream coverage uniformity.
3. End repair, A-tailing, and adapter ligation
Fragments are repaired to create blunt ends, followed by the addition of adenine overhangs (A-tailing) to enable T-overhang adapter compatibility. Platform-specific sequencing adapters are then ligated to each fragment, often carrying a unique index or barcode sequence that “tags” each sample. This enables samples to be pooled together, sequenced in a single run, then computationally demultiplexed post-sequencing — a practice that substantially reduces per-sample DNA sequencing costs.1
4. Size selection, cleanup, and PCR amplification
Size selection (via magnetic beads, columns, or gel) removes adapter dimers and off-target fragments, improving library quality before amplification. PCR enriches adapter-ligated fragments to achieve sufficient yield for sequencing. However, as excessive amplification introduces PCR bias, a careful balance between yield and library complexity must be maintained.
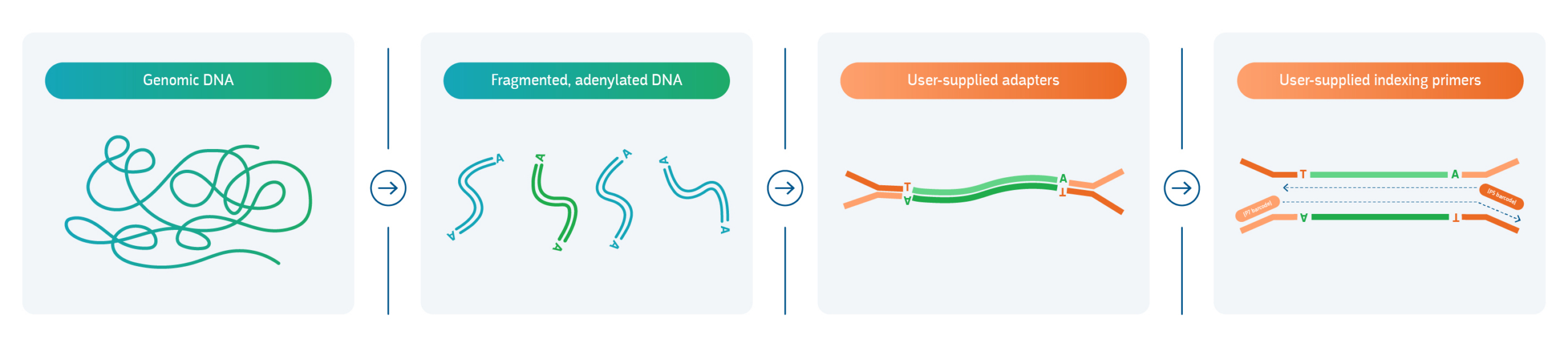 Figure 1. The general workflow for DNA library preparation.
Figure 1. The general workflow for DNA library preparation.
Common challenges in NGS library preparation
Even experienced labs encounter obstacles in generating high-quality NGS libraries, particularly when working with non-standard sample types.
Sample degradation
DNA or RNA from ancient, forensic, or FFPE samples is often degraded and the amount of usable template is often quite low. These attributes make it difficult to create libraries with optimal fragment size distributions and even coverage, leading to sequence dropout. These samples are especially costly when repeat analysis is required.
Low input quantity
Limited starting material often requires more PCR cycles to generate sufficient input for sequencing. However, increasing PCR cycles can increase the risk for amplification bias, leading to lower library complexity and a lowered ability to detect rare sequences. This is of particular concern when multiplexing libraries—it’s even more challenging to detect rare sequences within a sample pool.
GC bias
Genomes with particularly high or low GC content are prone to preferential amplification of certain sequences, reducing library diversity and skewing coverage uniformity and quantification accuracy.
These challenges are common for researchers in museomics, environmental surveillance, pathogen genomics, and agrigenomics where working with complex samples is the norm, but NGS library preparation is evolving to improve data recovery from complex sample types, helping researchers extract fresh insights from their samples.
Solutions: choosing the right NGS library preparation kit
Not all NGS library preparation kits are created equal. Quality kits are engineered for these challenges: advanced enzyme formulations that minimize GC bias, protocols optimized for low-input and degraded samples, and streamlined workflows that reduce error and hands-on time.
Daicel Arbor Biosciences NGS library preparation kits are designed for sample flexibility, workflow efficiency, and superior performance. Compatible with 1–500 ng inputs of high molecular weight or degraded dsDNA, they perform across diverse sample types that include non-model organisms, ancient specimens, environmental metagenomes, and clinical samples. Enzymatic fragmentation generates high-complexity libraries, and the kits are fully compatible with Daicel Arbor Biosciences NGS library preparation kits perform with the reliability and reproducibility needed to advance research and promote discovery.
When to leverage expert NGS services
For projects where in-house library preparation isn’t feasible, or where sample complexity demands specialized expertise, professional NGS services offer a solution. Outsourcing eliminates infrastructure barriers, improves reproducibility, and frees your team to focus on data analysis and experimental design. This is especially valuable when scaling for large studies or working with rare, irreplaceable specimens.
Daicel Arbor Biosciences offers support with expert library preparation services. Whether you’re processing degraded specimens, low-concentration samples, or running a large-scale research program, you can advance your research with a solution tailored to your needs.
Conclusion
Quality NGS library preparation is the foundation of reliable sequencing data.1 Whether you’re working with pristine genomic DNA or a heavily degraded sample, the chemistry and workflow you choose directly shape what you can learn from your data.
Ready to get started?
Explore Daicel Arbor Biosciences NGS Library Preparation Kits for your in-house workflows or discover Library Preparation Services that can support your most challenging projects. Contact us to discuss your specific research needs.
References
- Head SR, Komori HK, LaMere SA, et al. (2014). Library construction for next-generation sequencing: Overviews and challenges. BioTechniques, 56(2):61–77. PMC4351865
- Sato MP, Ogura Y, Nakamura K, et al. (2020). Library preparation for next generation sequencing: A review of automation strategies. Biotechnology Advances, 41:107537. ScienceDirect
 Linkedin
Linkedin Place order
Place order Quote request
Quote request Support
Support