Overview
myBaits® Custom Methyl-Seq kits for targeted methylation NGS are the perfect solution for human epigenetics research, enabling efficient deep sequencing of methylated and non-methylated genomic regions of interest in hundreds of samples per sequencing run.
With Daicel Arbor’s innovative methylation sequencing probe design algorithm and optimized hybridization capture protocol, you can efficiently enrich target genes and markers of interest from bisulfite- or enzymatically-converted methyl-seq NGS libraries. This significantly reduces per-sample sequencing costs compared to whole-genome bisulfite sequencing (WGBS) while preserving the essential strand-specific methylation signals.
myBaits kits can be easily integrated into your own research workflows—or paired with our customizable Sequencing or Bioinformatics services for a seamless NGS solution.
Affordable and scalable—Our proprietary oligo synthesis platform and unique approach to probe design for enzymatic- or bisulfite-converted DNA allows flexible production of high-quality methyl-seq enrichment probes at competitive pricing.
Flexible design—Our proprietary probe design algorithm is compatible with both novel and known methylation signals to craft efficient targeted methyl-seq capture probes, for human or other species.
Broad range of panel sizes—myBaits kits can accommodate any number of target loci, and offer an assortment of kit reaction sizes to accommodate any number of project samples.
Access to expertise—Our team of expert scientists can provide complimentary bait and project design assistance.
Easy to use—Easy-to-follow, dedicated methyl-seq enrichment protocol delivers consistently reproducible results, making myBaits kits ideal for new or expert users.
Convenient kits—Each myBaits kit includes probes and hybridization/washing reagents.
myBaits kits are for research use only and are not validated for diagnostic or therapeutic purposes.
Performance
DNA methylation occurs at cytosines within CpG and other dinucleotides and is a primary epigenetic mechanism controlling a variety of biological processes, especially transcriptional regulation of cell development and various diseases, including cancer.
Whole genome bisulfite sequencing (WGBS) has become the gold standard in measuring methylation status genome-wide, as it enables methylation signatures to be discovered in a wide range of sample types. By targeting thousands to millions of bases of relevant genomic regions in a single capture reaction, myBaits Custom Methyl-Seq offers an extremely cost-effective solution for enabling deep epigenetic sequencing for large numbers of samples. (Download app note)
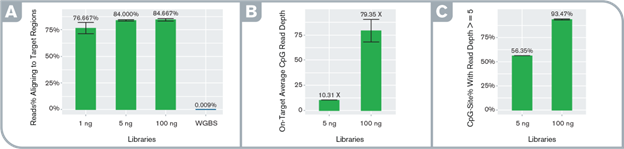
High specificity and orders of magnitude target enrichment. Performance metrics: (A) percentage of on-target reads in enriched and WGBS libraries, (B) average per-site depth of coverage at on-target CpG sites at an even subsample of 950K read-pairs in the enriched libraries, (C) percentage of on-target CpGs with read depth ≥5 at the even subsample in the enriched libraries. Libraries were prepared with the Swift Biosciences Accel-NGS Methyl-Seq Library Kit with varying input amounts and captured with the Daicel Arbor Biosciences myBaits Custom Methyl-Seq system targeting putative promoter regions of 50 cancer-associated genes (total target size ~100Kb).
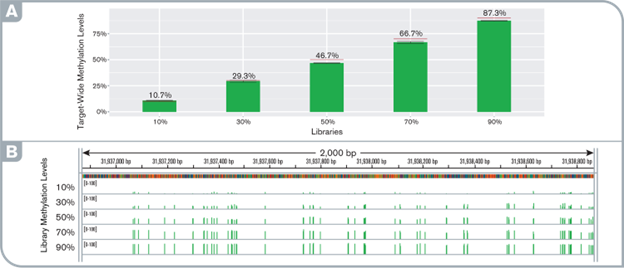
High sensitivity and fidelity of targeted methylation detection at any methylation levels
(A) target-wide methylation levels in pre- and post-capture libraries. Red lines indicate the simulated level. (B) IGV screenshot showing the uniform distribution of methylation levels along the gene STK12 promoter locus observed with the capture libraries at various methylation levels.
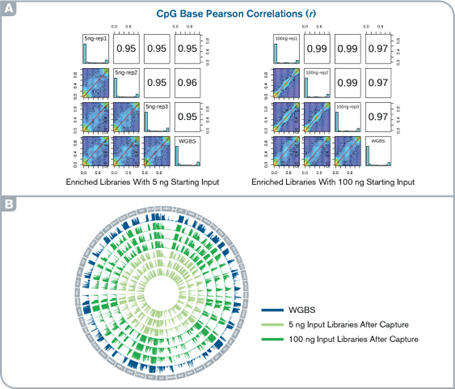
High accuracy and reproducibility of targeted methylation capture
(A) Histograms and scatter plots show the comparison of CpG methylation levels across the target regions between WGBS and capture libraries and among replicates. (B) Circos plot illustrating methylation patterns for the entire target regions in capture and WGBS libraries.
myBaits kits are for research use only and are not validated for diagnostic or therapeutic purposes.
Research Applications
myBaits Custom Methyl-Seq kits are ideal for enriching NGS libraries built from bisulfite or enzymatically converted DNA for:
- Re-sequencing methylated regions
- Epigenetics research
- Novel methylation patterns
- Cancer research
- Cell-free DNA
- And more
Featured publication:
Buckley et al. (2022). Targeted DNA methylation from cell-free DNA using hybridization probe capture. NAR Genomics and Bioinformatics 4(4): lqac099
myBaits kits are for research use only and are not validated for diagnostic or therapeutic purposes.
Resources

Product Literature
Poster
A universal targeted sequencing system for any high-throughput sequencing platform

myBaits kits are for research use only and are not validated for diagnostic or therapeutic purposes.
FAQs
Yes. Specific recommendations for library pooling for co-enrichment with myBaits Custom Methyl-Seq kits can be found in the applicable myBaits manual.
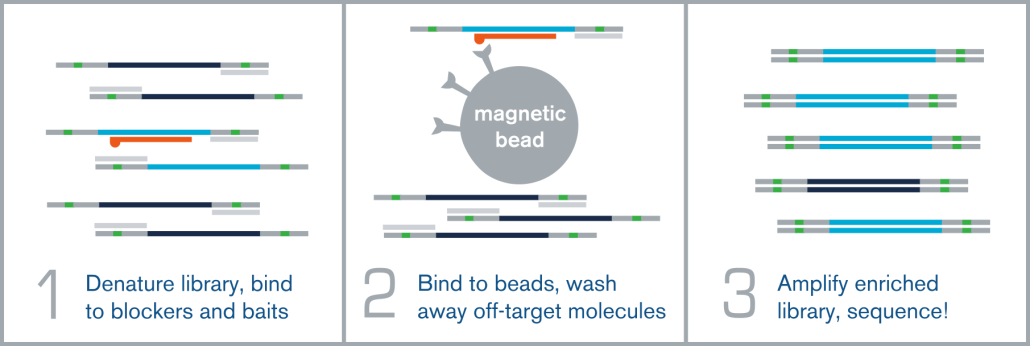
Hybridization capture is integrated into the overall NGS workflow immediately before sequencing on an NGS platform, such as Illumina. A fully sequenceable, barcoded/indexed NGS library is made from bisulfite or enzymatically-converted DNA. A pool of these multiple barcoded NGS libraries is then denatured, and allowed to anneal to complementary target-specific biotinylated probes/baits. These bait:library complexes are then bound to streptavidin-coated magnetic beads via the biotin on the probes, which are washed to remove non-specifically bound molecules. The remaining “enriched” library molecules are then released from the baits and amplified before sequencing.
Note! You may know the “hybridization capture” technique by another name, such as:
- Target enrichment
- Target capture
- Probe capture
- Exon capture
- Capture sequencing / sequence capture
- Hybridization sequencing / hyb-seq
Use myBaits Custom Methyl-Seq with PCR-amplified and amplifiable NGS libraries generated from bisulfite- or enzymatically-converted nucleic acids in which non-methylated cytosines have been converted to uracils, and following PCR amplification these positions are now thymines. Compatible formats include Illumina® TruSeq®-style, Illumina Nextera® Flex-style, Ion Torrent®, or other libraries with universal adapter priming sites. Do NOT use myBaits with PCR-free libraries; additionally, myBaits are incompatible with libraries made using original Nextera or Nextera XT library preparation kits, or any library type containing biotin. Dual-indexed libraries are strongly recommended to reduce the hazard of mis-indexing induced by PCR jumping events. The applicable myBaits manual provides detailed protocol instructions for enriching libraries intended for methylation sequencing.
If you are using a never-before-tried library prep protocol to pair with your myBaits kit, we recommend that you first perform some total library (shotgun) sequencing before doing myBaits enrichment. This is important in order to verify that your chosen library prep protocol/kit generates libraries of sufficient complexity and minimal bias in your hands, otherwise you may experience poor target capture results. High quality libraries are absolutely essential for achieving a successful target capture project.
myBaits hybridization capture kits include:
- Biotinylated RNA probes, with sequences corresponding to your custom design or a predesigned catalog option
- Hybridization and wash reagents
- For myBaits Custom kits, optional custom probe design informatics service (= expert bioinformaticians design and filter bait sequences and provide summary report and recommendations)
You will receive enough probes and reagents for performing the stated number of individual capture reactions of your kit size (e.g., 16 reactions) according to our current protocol. Please note that there are some additional reagents and equipment you will need to supply in order to perform a myBaits capture. Please review the list of required materials in the applicable myBaits manual to make sure you have everything you need before starting your experiments.
We also offer reagents for preparing libraries from DNA samples in advance of performing the myBaits hybridization capture step. Please visit Library Prep Kit for myBaits for more information.
If you are looking to outsource your project to a full-service laboratory and bioinformatics services group, please visit our myReads NGS laboratory and bioinformatics services page for more information about our comprehensive targeted sequencing service options (library preparation, target capture, next-generation sequencing, and optional analysis).
In the context of hybridization capture for targeted next generation sequencing, we use the terms interchangeably. Some fields prefer one term over the other, so we use both terms.
Turnaround time for myBaits targeted next generation sequencing kits varies based on design.
For new myBaits Custom baitset designs, the estimated manufacturing lead time is ~3-4 weeks minimum, starting from when your order is received and you have approved the final design. In addition, please consider that if you utilize our included bait design services, we will typically be in correspondence for an additional upfront period (up to several weeks) regarding a design before manufacturing can begin. Please also remember to accommodate any additional time for your collaborators to approve the final design, if applicable.
For myBaits Expert (catalog) kits or reorders of myBaits Custom kits with designs previously manufactured by Daicel Arbor Biosciences, the estimated manufacturing lead time is up to ~1-2 weeks from the time an order is received.
All myBaits kits include a specific protocol for their use as well as almost all of the reagents required to deploy them. In the manual, you will find the complete list of required supplies (reagents and equipment) that you will need in order to perform the captures.
Please see the applicable myBaits manual for detailed protocol instructions for enriching from Standard, High-Sensitivity, Long-Insert, or other specialty target/sample types.
Hybridization capture is integrated into the overall next generation sequencing workflow immediately before sequencing on an NGS platform, such as Illumina. A fully sequenceable, barcoded/indexed NGS library (or pool of multiple libraries) is denatured, and allowed to anneal to complementary target-specific biotinylated probes/baits. These bait:library complexes are then bound to streptavidin-coated magnetic beads via the biotin on the probes, which are washed to remove non-specifically bound molecules. The remaining “enriched” library molecules are then released from the baits and amplified before sequencing.
Note! You may know the “hybridization capture” technique by another name, such as:
- Target enrichment
- Target capture
- Probe capture
- Exon capture
- Capture sequencing / sequence capture
- Hybridization sequencing / hyb-seq
- Hybridization capture / hyb-cap
Specific recommendations for per-library input mass for different enrichment project types can be found in the applicable myBaits manual.
Target capture necessarily requires subjecting your libraries to a bottleneck, wherein target molecules are captured and therefore enriched, and non-target molecules are therefore removed. To have sufficient unique molecules for good sequencing coverage of your targets, successful captures DEPEND on the input of sufficiently complex libraries.
For best results, it is recommended that only amplified (non-PCR-free) NGS libraries are used for target capture. This provides multiple copies of each starting template molecule, increasing the chance of each individual molecule getting enriched. However if you need more starting material to reach the recommended amount, it is generally preferable to generate more library from fresh genomic DNA or a new batch of indexed library, rather than through extra amplification. This is because while some amplification is good, over-amplification risks reducing the observable complexity of your libraries through the uneven action of PCR bias, as some molecules will become relatively more abundant while others become rare. This is also true for manipulating your libraries after capture: amplify your post-capture libraries the minimum number of cycles necessary to reach the molarity required by your sequencing facility.
The applicable myBaits manual covers some common technical questions and troubleshooting topics at the end of each protocol. Please read through the relevant section first as it may answer your question. If you still have an issue, please contact us via email at techsupport_at_arbor.daicel.com or reach out to your most recent contact person for assistance.
When ordering your myBaits kit, please indicate the sequencing library configuration you intend to enrich. The standard adapter blocking reagent provided with the kit (Block X) is compatible with Illumina® TruSeq®-style or Nextera®-style libraries with single 6-12 bp or dual 6-12 bp indexing. These options cover the vast majority of currently available commercial library preparation systems intended for sequencing on any Illumina platform.
For different adapter configurations than those described above, we recommend ordering Custom IDT® xGen® Blocking Oligos. At a concentration of 1 μg/μL, custom adapter-blocking oligos can be used in lieu of myBaits Block X.
If you are not certain, or later decide to change your library prep kit, please contact us so we can instruct you on how to obtain the correct blocking oligos.
Yes! Our expert myReads team provides a range of in-house NGS services, including library preparation, target capture with myBaits, high-throughput sequencing, and optional bioinformatics analysis. Visit the Sequencing Services page to learn more about our comprehensive laboratory and sequencing service options!
Yes! As long as we receive written permission from the original designer(s) (if it is not your kit and the bait sequences are not publicly available), you can re-order any past design that has been manufactured by Daicel Arbor Biosciences. We can usually provide such re-orders within ~1-2 weeks of ordering. This ensures consistency in your targeted next-generation sequencing workflow.
Sequence Submission Guidelines [PDF]
Please gather your target sequences in FASTA format or as genomic coordinates according to our guidelines, and contact us with details of your project. Our team will provide you with an estimated panel size as soon as possible based on your provided information. Please let us know upfront if there is a specific panel size in which your design should be constrained (e.g. not more than 60,000 probes) so that together we can adjust your design/estimate accordingly. Otherwise, our experts will determine the best size of panel based on your targets and project configuration.
myBaits Custom kits have frequently achieved high on-target percentages for a wide range of applications. However since it is not possible to predict the behavior of new baitsets (e.g. on-target percentage, unique read depth, and evenness of coverage) without experimental test data, and knowledge of your experimental parameters, we are unable to provide specific predictions for downstream sequencing performance. Factors such as the overall size and GC content of the bait sequences, the sequence divergence between baits and targets, the quality of your NGS libraries, and the sequencing depth will also have significant impacts on post-enrichment outcomes.
If sequencing efficiency is critical to your project, best practice for optimizing new target capture designs is to perform a pilot test to determine the behavior of the baitset under your chosen conditions and with your samples, and adjust parameters such as sequencing depth, hybridization stringency, or number of capture rounds accordingly. For example, to maximize your on-target percentage, you could consider making upfront protocol adjustments such as performing two consecutive rounds of capture, as long as you are working with sufficiently high-quality, complex libraries.
Singleton and/or short stretches of N’s will be replaced with T’s to facilitate bait design in these regions. Longer stretches (e.g 10+ N’s) will be skipped over during bait placement.
Ambiguities (e.g. Y/M/R/S/W/K) are allowed, but will be replaced by ONE random candidate base for manufacturing, since we only synthesize A/T/C/G bases (no mixed bases). The hybridization capture system tolerates multiple mismatches between probe:target molecules. However, sequences that contain on average >5-7% ambiguous bases are not recommended. If you are providing consensus sequence(s) generated from a common locus/gene source (e.g. the same gene from multiple genomes, or multiple alleles of a target gene), please provide the original individual sequences. Our informatics experts can remove redundant/similar regions during the design process to ensure all variants are sufficiently represented while minimizing overall unique bait count.
We are pleased to provide as much bait design advice and assistance as possible. However we are unlikely to be sufficiently knowledgeable in your particular field as to help you pick the specific genes/targets for your project. Whether this is your first NGS project and/or you are an experienced genetics researcher, we always recommend that you choose your targets in collaboration with your full research team, especially your bioinformatician(s), so that your kit design is as robust as possible.
Some general suggestions appropriate for many projects would be to exhaustively survey the literature for your organism(s), and consider including neutral and/or control loci in addition to specific targets of interest. You should include enough loci and/or SNPs to draw significant conclusions within the number of specimens that you plan to survey. You should make sure that you have thoroughly evaluated your bait design before proceeding with your kit order.
If you are beginning a completely new project, you may wish to order the smallest number of reactions upfront, and place a reorder for a larger number of reactions once you have tested the design. However please note that any changes to your design (adding or removing baits) would be ordered as a fully new custom kit, which may have a longer delivery time than a reorder of a previous design.
myBaits kits are for research use only and are not validated for diagnostic or therapeutic purposes.


 Linkedin
Linkedin Place order
Place order Quote request
Quote request Support
Support