
The myTXTL Linear DNA Expression Kit is based off our myTXTL Sigma 70 Master Mix Kit, but has been further engineered to efficiently produce soluble and membrane proteins using linear DNA templates without the need for nuclease inhibitors like GamS. Simply add linear DNA template to the optimized master mix to begin protein synthesis.
All P70 promoters originate from the lambda phage promoter for the repressor Cro with its two operator sites and are specific to the E. coli sigma factor 70. They differ in strength (P70a > P70d > P70b > P70c) due to mutations that were introduced at -35 and/or -10 regions.
We recommend setting the myTXTL GamS Protein concentration in a myTXTL reaction at 10 uM (myTXTL GamS stock solution is provided at 150 uM).
We offer a range of fluorescent and non-fluorescent label options for myTags Custom labeled in situ hybridization probes. Please contact us for current available label options.
The available label options enable compatibility with most microscopes or other visualization instruments, as well as enabling detecting multiple targets within the same sample by using spectrally distinct fluorophores for individual probesets.
We may be able to accommodate other labeling options, please contact us for availability.
The number of assays per library depends on a number of factors including the probe density of your library, the size of your target region, the number of probes in the library, and the FISH protocol. Generally we recommend starting with 10pmol of labeled probes per standard FISH slide and then modifying the input amount based on the initial results.
The latest recommended protocols for both labeled and immortal probe libraries can be found in the Resources tab, but in general, myTags in situ hybridization libraries are compatible with most FISH protocols. Please contact us for specific recommendations.
We can often accommodate customer-designed probes into the myTags labeling framework. Please contact us for recommendations on the design parameters and other information before designing your probe sequences.
Yes, we can synthesize immortal probe libraries that can be labeled using the Oligopaints labeling method. Please note these probe libraries are not compatible with the standard myTags labeling protocol due to sequence requirements of the Oligopaints method.
We generally ask for up to 3-4 weeks after an order is placed to ship myTags libraries.
We recommend using the myTags labeling protocol with myTags immortal libraries for perfroming the labeling process in your own lab. If you would like to use a different protocol, one of our scientists would be happy to provide assistance to ensure success.
Probes are available in 2 configurations to detect both the positive (+) strand and negative (-) strand sequences:
Positive (+) strand probes– SARS-CoV-2 is a class IV RNA virus with an ssRNA genome (positive strand). The probes to detect the (+) strand will hybridize to full-length genomic (+) strand RNA as well as subgenomic (+) strand RNAs made during replication.
Negative (-) strand probes– Detection of the (-) strand is a hallmark of active viral replication. The probes to detect the (-) strand will hybridize to the full-length (-) strand as well as subgenomic (-) strand RNAs. In experimental applications where viral infection is not controlled, additional confirmation of SARS-CoV-2 presence via PCR is recommended.
NOTE: The positive (+) and negative (-) strand probes are not designed to be used in the same hybridization experiment. If co-hybridization is required for your experimental design, please contact us for a customized probe solution.
myTags Expert SARS-CoV-2 probes are available in ready-to-use labeled formats, labeled with Alexa-488, ATTO-550, or ATTO-647N fluorescent tags. For experiments that need a brighter signal, the 3X fluorescent tag upgrade is recommended, which can aid detection of lower cellular viral RNA concentrations at earlier experimental time points.
Yes. Due to the manufacturing process, there might be a small pellet visible. It is critical that you resuspend this pellet back into the myTXTL Master Mix completely before aliquoting it to set up your myTXTL reaction(s).
Yes, myTXTL Linear DNA Expression Kit allows the use of both, linear and circular/plasmid templates.
Sample handling and storage is mainly determined by the stability of your molecule of interest (protein, DNA, RNA) and thus optimal conditions may need to be evaluated. But to ensure sample integrity, we would recommend to either process the myTXTL reaction immediately after performing the incubation or store it at ≤ -20 °C.
Limit the number of freeze-thaw-cycles as much as possible. Our studies have indicated that up to five freeze-thaw-cycles do not negatively influence protein production efficiency of the myTXTL Master Mix when using flash freezing in liquid nitrogen and subsequent -80C storage.
Yes, please see examples in the Publications section; filter for myTXTL. Please note, that every gene circuit should start with a σ70-specific promoter like P70a.
Most importantly, the excitation and emission wavelength should match the fluorescence properties of deGFP/eGFP (e.g. λEm 488 nm, λEx 535 nm). Other reader settings such as reading mode, integration time and gain value should be chosen under consideration of high well-to-well fluorescence reading reproducibility.
Yes. For all plasmids containing the lambda phage promoter (P70a, P70b, P70c, P70d) it is extremely crucial to use E. coli KL740 as the transformation strain. When cultivated below 30°C, this strain over-expresses the lambda phage repressor protein Cl857 that represses P70 promoters, thus ensuring high transformation efficiency and plasmid stability. KL740 can be purchased from E. coli Genetic Stock Center (Yale) [CGSC#: 4382] or from Daicel Arbor. For all other plasmids, a standard laboratory E. coli cloning strain like JM109 or DH5alpha is sufficient.
Yes, Daicel Arbor Biosciences has an NGS services division adept at preparing and working with ancient DNA libraries. Learn more about our myReads NGS laboratory and sequencing service options, and contact us today to discuss your NGS project goals with our team of ancient DNA experts.
Yes. Specific recommendations for library pooling for co-enrichment with myBaits Custom Methyl-Seq kits can be found in the applicable myBaits manual.
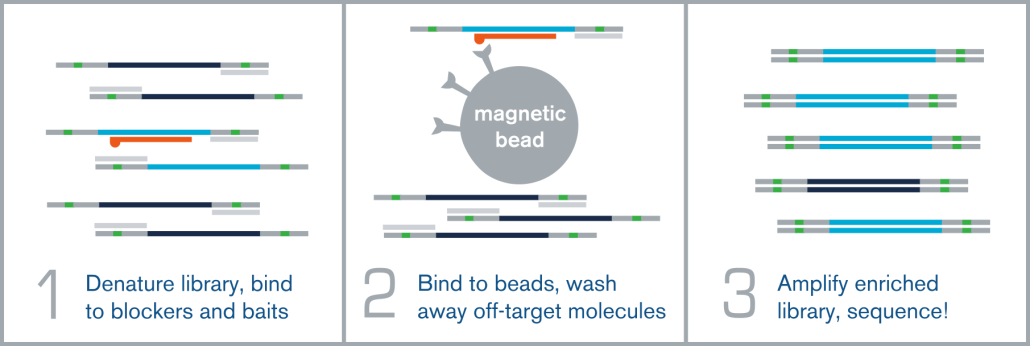
Hybridization capture is integrated into the overall NGS workflow immediately before sequencing on an NGS platform, such as Illumina. A fully sequenceable, barcoded/indexed NGS library is made from bisulfite or enzymatically-converted DNA. A pool of these multiple barcoded NGS libraries is then denatured, and allowed to anneal to complementary target-specific biotinylated probes/baits. These bait:library complexes are then bound to streptavidin-coated magnetic beads via the biotin on the probes, which are washed to remove non-specifically bound molecules. The remaining “enriched” library molecules are then released from the baits and amplified before sequencing.
Note! You may know the “hybridization capture” technique by another name, such as:
- Target enrichment
- Target capture
- Probe capture
- Exon capture
- Capture sequencing / sequence capture
- Hybridization sequencing / hyb-seq
Use myBaits Custom Methyl-Seq with PCR-amplified and amplifiable NGS libraries generated from bisulfite- or enzymatically-converted nucleic acids in which non-methylated cytosines have been converted to uracils, and following PCR amplification these positions are now thymines. Compatible formats include Illumina® TruSeq®-style, Illumina Nextera® Flex-style, Ion Torrent®, or other libraries with universal adapter priming sites. Do NOT use myBaits with PCR-free libraries; additionally, myBaits are incompatible with libraries made using original Nextera or Nextera XT library preparation kits, or any library type containing biotin. Dual-indexed libraries are strongly recommended to reduce the hazard of mis-indexing induced by PCR jumping events. The applicable myBaits manual provides detailed protocol instructions for enriching libraries intended for methylation sequencing.
If you are using a never-before-tried library prep protocol to pair with your myBaits kit, we recommend that you first perform some total library (shotgun) sequencing before doing myBaits enrichment. This is important in order to verify that your chosen library prep protocol/kit generates libraries of sufficient complexity and minimal bias in your hands, otherwise you may experience poor target capture results. High quality libraries are absolutely essential for achieving a successful target capture project.
Use myBaits with PCR-amplified and amplifiable NGS libraries, including Illumina TruSeq® -style, Illumina Nextera® Flex-style, Ion Torrent, or other libraries with universal adapter priming sites. It is NOT recommended to use myBaits with PCR-free libraries. Additionally, myBaits are incompatible with libraries made using original Nextera or Nextera XT library preparation kits, or any library type containing biotin. Dual-indexed libraries are strongly recommended to reduce the hazard of mis-indexing induced by PCR jumping events. The applicable myBaits manual provides detailed protocol instructions for enriching libraries for sequencing on short- and/or long-read platforms (e.g. PacBio® or Oxford Nanopore Technologies®).
If you are using a never-before-tried library prep protocol to pair with your myBaits kit, we recommend that you first perform some total library (shotgun) sequencing before doing myBaits enrichment. This is important in order to verify that your chosen library prep protocol/kit generates libraries of sufficient complexity and minimal bias in your hands, otherwise you may experience poor target capture results. High quality libraries are absolutely essential for achieving a successful target capture project.
Provided below are a list of companies that sell NGS library prep kits that are known to be compatible with myBaits. This is NOT an exhaustive list; there are many other unlisted options that are also compatible with myBaits. Also, kits on this list may not necessarily be appropriate for your samples. NGS library prep is not “one size fits all”; different factors such as sample type, DNA input amount, genome complexity, and sequence composition may influence the type of library prep kit that would be best for your application. For example, low input, degraded, and/or damaged DNA templates may require special handling (see below) and/or modifications to commercial kits.
Contact these and other manufacturers to learn about your options and find what works best for your samples and project needs:
- Biosearch / Lucigen
- Claret Bioscience
- Illumina
- New England Biolabs
- Kapa Biosystems
- PerkinElmer / Bioo Scientific
- Rubicon Genomics / Takara
- Swift Biosciences / IDT
 Linkedin
Linkedin Place order
Place order Quote request
Quote request Support
Support