
To produce a Custom WGE panel, a high-purity, high-molecular weight genomic DNA (gDNA) sample is required to be shipped to Arbor’s manufacturing facility in Ann Arbor, MI, USA.
If you do not have access to the appropriate gDNA, for some organisms Arbor may be able to provide the additional service of acquiring the gDNA sample(s) from a third-party vendor.
Custom gDNA sourcing service is only upon prior arrangement and approval by Arbor. This service is only available for species whose genomic DNAs are available from vendors supplying the US. The gDNA sample(s) must be available for Arbor to purchase in sufficient quantity as a commercial entity without any special licenses, acquisition or use permits, or memberships.
Please contact us to provide specific details on the gDNA source(s)/species that you are interested in, and we will review the request to determine feasibility and pricing.
Target capture necessarily requires subjecting your libraries to a bottleneck, wherein target molecules are captured and enriched, and non-target molecules are therefore removed. To have sufficient unique molecules for good sequencing coverage of your targets, successful captures DEPEND on the input of sufficiently complex libraries. For this reason, for libraries with a significant non-target component (e.g., ancient, forensic, or environmental samples), and especially for WGE captures with a very large full nuclear genome target size, we strongly recommend maximizing the target component in each capture by using as much input library as possible (up to 2 µg+), and consider two rounds of capture for higher percentage of reads on-target.
For best results, it is recommended that only amplified (non-PCR-free) NGS libraries are used for target capture. This provides multiple copies of each starting template molecule, increasing the chance of each individual molecule getting enriched. However if you need more starting material to reach the recommended amount, it is generally preferable to generate more library from fresh genomic DNA or a new batch of indexed library, rather than through extra amplification. This is because while some amplification is good, over-amplification risks reducing the observable complexity of your libraries through the uneven action of PCR bias, as some molecules will become relatively more abundant while others become rare. This is also true for manipulating your libraries after capture: amplify your post-capture libraries the minimum number of cycles necessary to reach the molarity required by your sequencing facility.
We generally do NOT recommend pooling multiple samples per capture reaction for very degraded and/or rare targets (e.g. ancient DNA), or for very large targets (e.g. a WGE baitset targeting a full nuclear genome). In fact, for highly degraded samples, you may achieve better results by performing multiple parallel WGE reactions per sample.
The myBaits WGE product line ONLY offers bait production directly from high quality genomic DNA precursor material. This gDNA must be made physically available to us in order to manufacture a myBaits WGE Custom kit (either extracted in your lab or sourced from a third-party gDNA supplier).
Daicel Arbor Biosciences does have the ability to design and synthesize synthetic customized bait oligo pools via our standard myBaits Custom DNA-Seq, RNA-Seq, and Methyl-Seq products. However the myBaits Custom synthetic oligo approach is generally not an option for targeting entire large nuclear genomes (e.g. from eukaryotic organisms such as plants or animals) due to prohibitively large numbers of baits that would be required. However if you are working with an organism that has a much smaller genome (e.g. one or more bacteria), or are interested in capturing only a portion of a much larger genome, then a myBaits Custom kit is likely the most effective option.
Please contact us with details about your project goals and budget, so we can discuss project options.
We offer complementary myTags Custom (F)ISH probe bioinformatic design service using our proprietary design algorithms for most types of (F)ISH projects. Please contact us with a brief description of your project, including the name of your study species, genomic coordinates, and any additional information.
Our experts will apply our advanced proprietary probe design algorithms to craft a custom probe design for your targeting needs, for any organism or application. Our scientific team has decades of experience designing custom probes for a wide variety of applications, including multi- or single locus/gene localization, chromosome painting, chromosomal indexing/barcoding, haplotyping, and more.
Only the highest specificity regions with minimal background noise and cross-reactivity for premium performance in any downstream experimental application would be selected for the probe design.
And with our transparent and straightforward scientific communication process, you always have full control and ownership over the oligo sequences for each and every myTags Custom probeset.
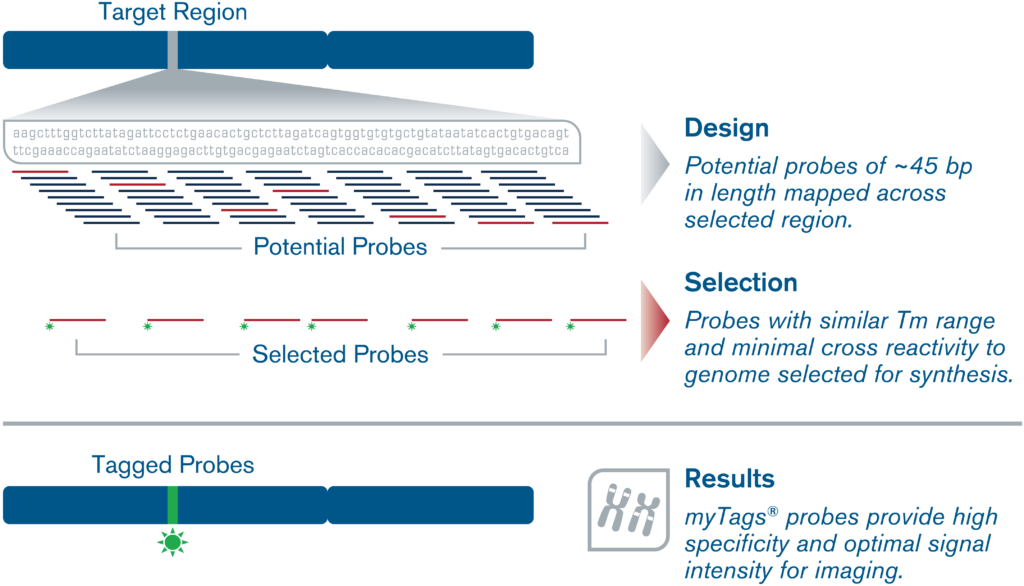
Generally we recommend probe densities between 3-10 probes per kilobase for target regions larger than 50kb. For target regions between 10-50kb, probe densities should be on the higher end of that range, and we may recommend using multiple fluorophores per probe to boost the signal.
We can work with any sequence to design probes. Please contact us with a brief description of your project, including the name of your study species, genomic coordinates, and any additional information.
Our experts will advise you on the most appropriate synthesis configuration for your desired project needs, which depends on the number of individual probe sequences required for your design. This is primarily driven by size of your target region(s), but also your experimental setup and goals.
You can choose to receive your individual probe pool(s) either as non-labeled (a.k.a. “immortal”) amplifiable substrate(s) ready for labeling in your lab, or receive as labeled, ready-to-use probe pool(s) for your experiments.
Regardless, all unique probesets are delivered individually, and include individual final probeset pool composition verification via next-generation sequencing.
myBaits hybridization capture kits include:
- Biotinylated RNA probes, with sequences corresponding to your custom design or a predesigned catalog option
- Hybridization and wash reagents
- For myBaits Custom kits, optional custom probe design informatics service (= expert bioinformaticians design and filter bait sequences and provide summary report and recommendations)
You will receive enough probes and reagents for performing the stated number of individual capture reactions of your kit size (e.g., 16 reactions) according to our current protocol. Please note that there are some additional reagents and equipment you will need to supply in order to perform a myBaits capture. Please review the list of required materials in the applicable myBaits manual to make sure you have everything you need before starting your experiments.
We also offer reagents for preparing libraries from DNA samples in advance of performing the myBaits hybridization capture step. Please visit Library Prep Kit for myBaits for more information.
If you are looking to outsource your project to a full-service laboratory and bioinformatics services group, please visit our myReads NGS laboratory and bioinformatics services page for more information about our comprehensive targeted sequencing service options (library preparation, target capture, next-generation sequencing, and optional analysis).
In the context of hybridization capture for targeted next generation sequencing, we use the terms interchangeably. Some fields prefer one term over the other, so we use both terms.
Turnaround time for myBaits targeted next generation sequencing kits varies based on design.
For new myBaits Custom baitset designs, the estimated manufacturing lead time is ~3-4 weeks minimum, starting from when your order is received and you have approved the final design. In addition, please consider that if you utilize our included bait design services, we will typically be in correspondence for an additional upfront period (up to several weeks) regarding a design before manufacturing can begin. Please also remember to accommodate any additional time for your collaborators to approve the final design, if applicable.
For myBaits Expert (catalog) kits or reorders of myBaits Custom kits with designs previously manufactured by Daicel Arbor Biosciences, the estimated manufacturing lead time is up to ~1-2 weeks from the time an order is received.
All myBaits kits include a specific protocol for their use as well as almost all of the reagents required to deploy them. In the manual, you will find the complete list of required supplies (reagents and equipment) that you will need in order to perform the captures.
Please see the applicable myBaits manual for detailed protocol instructions for enriching from Standard, High-Sensitivity, Long-Insert, or other specialty target/sample types.
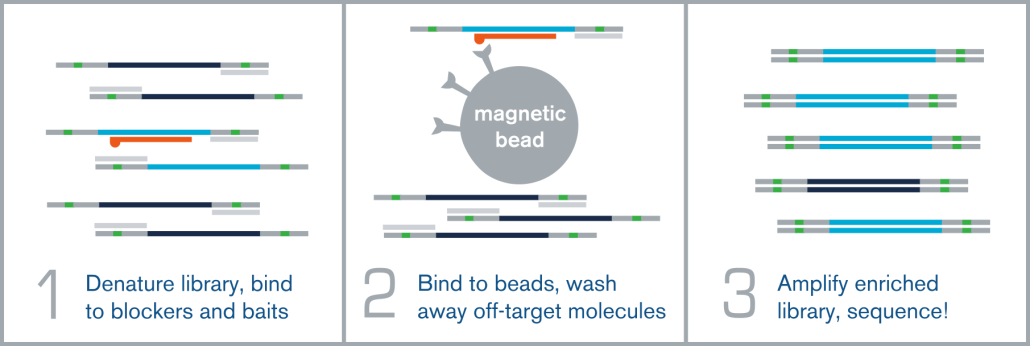
Hybridization capture is integrated into the overall next generation sequencing workflow immediately before sequencing on an NGS platform, such as Illumina. A fully sequenceable, barcoded/indexed NGS library (or pool of multiple libraries) is denatured, and allowed to anneal to complementary target-specific biotinylated probes/baits. These bait:library complexes are then bound to streptavidin-coated magnetic beads via the biotin on the probes, which are washed to remove non-specifically bound molecules. The remaining “enriched” library molecules are then released from the baits and amplified before sequencing.
Note! You may know the “hybridization capture” technique by another name, such as:
- Target enrichment
- Target capture
- Probe capture
- Exon capture
- Capture sequencing / sequence capture
- Hybridization sequencing / hyb-seq
- Hybridization capture / hyb-cap
Specific recommendations for per-library input mass for different enrichment project types can be found in the applicable myBaits manual.
Target capture necessarily requires subjecting your libraries to a bottleneck, wherein target molecules are captured and therefore enriched, and non-target molecules are therefore removed. To have sufficient unique molecules for good sequencing coverage of your targets, successful captures DEPEND on the input of sufficiently complex libraries.
For best results, it is recommended that only amplified (non-PCR-free) NGS libraries are used for target capture. This provides multiple copies of each starting template molecule, increasing the chance of each individual molecule getting enriched. However if you need more starting material to reach the recommended amount, it is generally preferable to generate more library from fresh genomic DNA or a new batch of indexed library, rather than through extra amplification. This is because while some amplification is good, over-amplification risks reducing the observable complexity of your libraries through the uneven action of PCR bias, as some molecules will become relatively more abundant while others become rare. This is also true for manipulating your libraries after capture: amplify your post-capture libraries the minimum number of cycles necessary to reach the molarity required by your sequencing facility.
The applicable myBaits manual covers some common technical questions and troubleshooting topics at the end of each protocol. Please read through the relevant section first as it may answer your question. If you still have an issue, please contact us via email at techsupport_at_arbor.daicel.com or reach out to your most recent contact person for assistance.
When ordering your myBaits kit, please indicate the sequencing library configuration you intend to enrich. The standard adapter blocking reagent provided with the kit (Block X) is compatible with Illumina® TruSeq®-style or Nextera®-style libraries with single 6-12 bp or dual 6-12 bp indexing. These options cover the vast majority of currently available commercial library preparation systems intended for sequencing on any Illumina platform.
For different adapter configurations than those described above, we recommend ordering Custom IDT® xGen® Blocking Oligos. At a concentration of 1 μg/μL, custom adapter-blocking oligos can be used in lieu of myBaits Block X.
If you are not certain, or later decide to change your library prep kit, please contact us so we can instruct you on how to obtain the correct blocking oligos.
Yes, please contact sales@arbor.daicel.com to add our subsampling service to your project.
We strongly discourage sending frozen plant samples because freeze-thaw cycles are terrible for DNA integrity. Also, we have found that lyophilization has large positive effects on yield and purity for plants. If you do submit frozen material, ensure that once it is frozen, it is not allowed to thaw.
We tested a wide variety of plastics and seals before making this recommendation. This is the combination that we have identified as capable of standing up to homogenization without leaking, so we do require these specific plastics to be used. Having trouble sourcing them? Contact genomics@arbor.daicel.com for help.
Yes. If they are submitted in our required plastics, you can add our lyophilization service to your project. If you would like us to format your samples correctly for lyophilization, we can do so for an additional fee.
Yes! Check out Appendix 2 of our Sample Preparation Guide. It has tips on how to get your plant subsamples into a plate. Tried our tips and still need help? You can add our subsampling service to your project.
No, due to the potential effects of upstream handling (which we cannot control), we cannot guarantee the output mass, purity, or integrity of extracted nucleic acids. Please see our NGS Services Policies.
If you make arrangements before placing your order, we may be able to return unused materials. However, we cannot guarantee there will be leftover material. We cannot return any samples for which an import permit was required to bring the samples into the USA.
Our extraction pilot services include the following:
Flexible sample submission: Send us 1–12 samples in any format, including fresh, frozen, or dried tissue, in tubes or plates.
Sample preparation: We’ll handle all necessary subsampling and reformatting, including lyophilization for plant samples when applicable.
Two extraction attempts: Each sample undergoes up to two extraction attempts. The first uses our best-fit protocol, and the second—if needed—incorporates protocol adjustments based on initial results.
Comprehensive quality control: You’ll receive a detailed QC report for each extraction attempt, including DNA yield (mass) and morphology analysis via TapeStation.
Post-extraction options: We can proceed directly to downstream myReads® workflows, return the extracted DNA to you (shipping fees apply), or store it for up to one year (long-term storage fees may apply).
Absolutely possible!
We’ve successfully worked with a wide variety of sample types — including ancient DNA, degraded specimens, fresh tissues, and samples containing challenging inhibitory compounds. Our experience spans across plant, animal, insect, and other complex biological materials, from ultra-low-input samples to high-volume extractions.
Even if your specific sample type is new to us, we’re always eager to take on unique challenges. We encourage you to explore our pilot extraction program, which allows us to test your samples using our protocols before you commit to a full project.
 Linkedin
Linkedin Place order
Place order Quote request
Quote request Support
Support