Evolutionary radiations are prominent and pervasive across many plant lineages in diverse geographical and ecological settings; in neotropical rainforests there is growing evidence suggesting that a significant fraction of species richness is the result of recent radiations. Understanding the evolutionary trajectories and mechanisms underlying these radiations demands much greater phylogenetic resolution than is currently available for these groups. The neotropical tree genus Inga (Leguminosae) is a good example, with ~300 extant species and a crown age of 2-10 MY, yet over 6kb of plastid and nuclear DNA sequence data gives only poor phylogenetic resolution among species. Here we explore the use of larger-scale nuclear gene data obtained though targeted enrichment to increase phylogenetic resolution within Inga. Transcriptome data from three Inga species were used to select 264 nuclear loci for targeted enrichment and sequencing. Following quality control to remove probable paralogs from these sequence data, the final dataset comprised 259,313 bases from 194 loci for 24 accessions representing 22 Inga species and an outgroup (Zygia). Bayesian phylogenies reconstructed using either all loci concatenated or a subset of 60 loci in a gene-tree/species-tree approach yielded highly resolved phylogenies. We used coalescent approaches to show that the same targeted enrichment data also have significant power to discriminate among alternative within-species population histories in the widespread species I. umbellifera. In either application, targeted enrichment simplifies the informatics challenge of identifying orthologous loci associated with de novo genome sequencing. We conclude that targeted enrichment provides the large volumes of phylogenetically-informative sequence data required to resolve relationships within recent plant species radiations, both at the species level and for within-species phylogeographic studies.
- Genomic or other dsDNA is enzymatically fragmented, end-polished, and adenylated
- User-supplied adapters are ligated to the end-repaired fragments
- Ligation products are purified with SPRI beads and then amplified and SPRI’d again
- Libraries are optionally pooled and taken to myBaits capture
In addition to the upstream library preparation steps, the Library Prep Kit for myBaits includes the reagents necessary for performing the post-capture amplification step of the myBaits protocol.
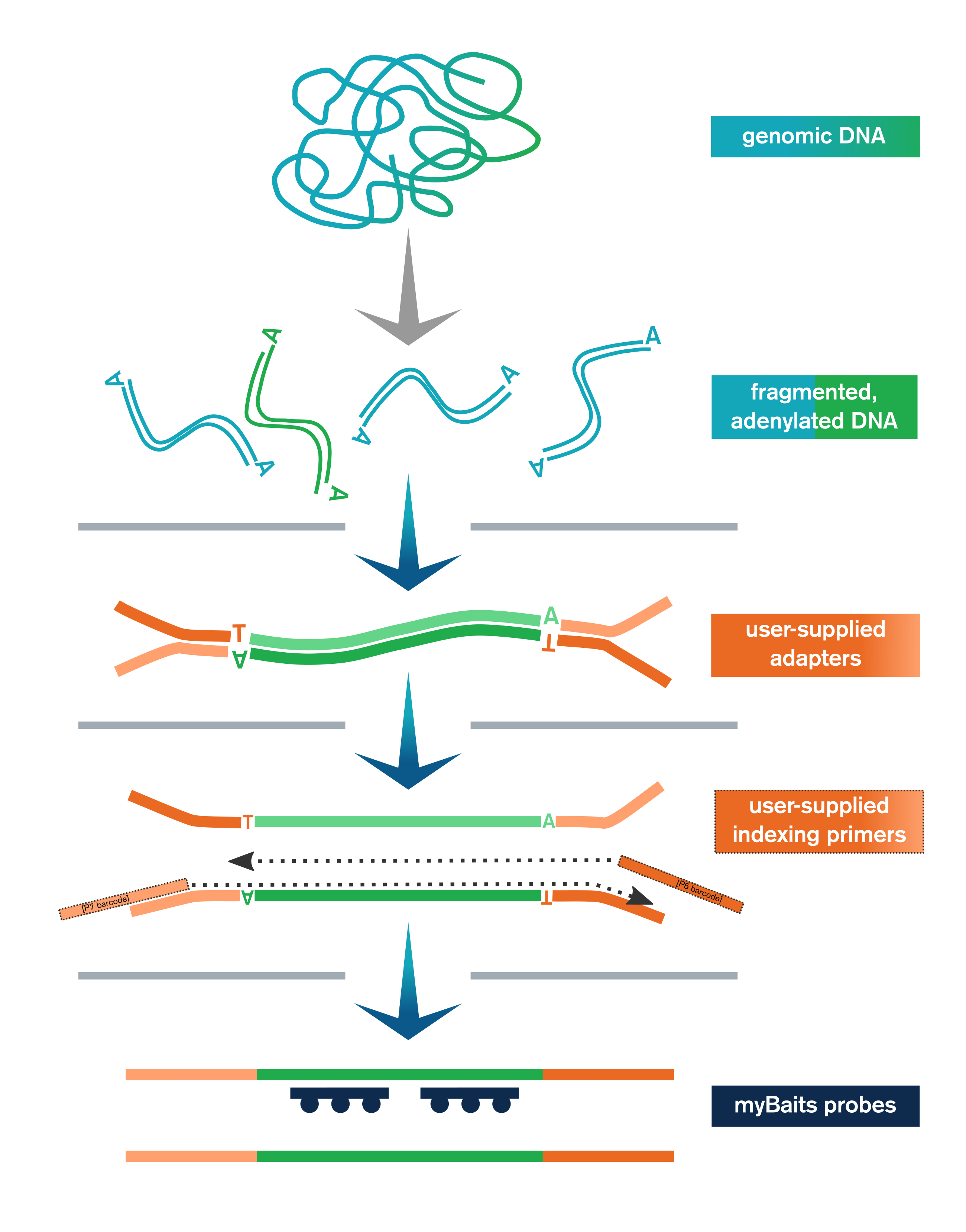
The Library Prep Kit includes reagents for end-polishing/A-tailing, enzymatic fragmentation, ligation of user-supplied adapters, purification, and amplification (with user-supplied indexing primers, or kit-supplied universal P5/P7 primers if using full-length adapters).
In addition, the kit also provides sufficient reagents for the amplification and purification steps at the end of the myBaits capture protocol (myBaits kit sold separately).
Full details of what is included in the kit and what reagents/equipment are required to complete the protocol can be found in the kit manual.
The Library Preparation Kit for myBaits is intended to be used for the preparation of NGS libraries from DNA samples prior to hybridization capture with myBaits.
Compatible DNA samples should be or have:
- 1-500 ng genomic or other dsDNA
- <= 0.1mM EDTA storage buffer
- No viscosity or coloration
- Any length distribution; see technical recommendations for selecting frag time
Libraries made with this protocol are directly compatible with downstream myBaits enrichment (or another hybridization capture system), or alternatively can be sequenced directly in order to generate non-enriched read data.
No. myBaits hybridization capture kits have always been– and will continue to be– compatible with any user-provided upstream NGS library preparation system that is appropriate for a given project.
However, for your convenience, we are now offering our own powerful kit for preparing NGS libraries from most types of DNA samples, which is relevant for many myBaits hybridization capture projects. This new product (Library Preparation Kit for myBaits) is intended to be used for the preparation of NGS libraries from dsDNA samples prior to hybridization capture with myBaits.
In addition to the upstream library preparation steps, the Library Prep Kit for myBaits includes the reagents necessary for performing the post-capture amplification step of the myBaits protocol.
The Library Prep Kit for myBaits is compatible with a wide range of pre-built or DIY adapters or primers. The adapters used must be T-overhang-containing double-stranded adapters. These can be either short (“stubby”) adapters OR full-length adapters that contain sample-specific barcodes. If using stubby adapters, indexing primers that add universal P5 and P7 priming sites are also required. See the kit manual for further technical information about adapters and primers.
We recommend using unique dual indexes. When selecting indexes, ensure that all libraries that you ever plan to co-enrich or co-sequence have unique index combos.
For your convenience, below are listed some commercially-available adapter and barcoding solutions designed for Illumina(R) short-read sequencing, but other vendors also supply compatible options.
Stubby adapters + indexing primers
- IDT
- xGen™ Stubby Adapter in 16 rxn (10005974) or 96 rxn (10005924)
- xGen™ UDI Primer Pairs in 8nt 16 rxn (10005975), 8nt Plate 1 (10005922), 10nt Plates 1-4 (10008052), 10nt Plates 1-8 (10008053), or 10nt Plates 1-16 (10008054)
- NEB
- NEBNext® Multiplex Oligos for Illumina® | 96 Unique Dual Index Primer Pairs – Set 1 (E6440S), Set 2 (E6442S), Set 3 (E6444S), Set 4 (E6446S), Set 5 (E6448S). (Note: Do not use the included “NEBNext Adaptor” unless you open the hairpin loop with USER treatment.)
Full-length adapters
- IDT
- xGen™ UDI-UMI Adapters in 16 rxn (10006914) or 96 rxn (10005903)
- NEB
- NEBNext® Multiplex Oligos for Illumina® | Unique Dual Index UMI Adaptors DNA – Set 1 (E7395S), Set 2 (E7874S), Set 3 (E7876S), Set 4 (E7878S)
The Library Prep Kit for myBaits is designed for input amounts from 1 to 500 ng. Please see the kit manual for technical recommendations.
Please see the kit manual for detailed technical recommendations on the enzymatic fragmentation step. In brief, the final length of your DNA after fragmentation will depend on the (1) the length of your starting genomic DNA sample and (2) your chosen fragmentation time.
myBaits hybridization capture can tolerate NGS library insert lengths from 50bp-10Kbp. However for most myBaits applications, users should aim to generate NGS libraries with ~300-500bp length inserts (~450-650bp length libraries).
 Twitter
Twitter Linkedin
Linkedin Place order
Place order Quote request
Quote request Support
Support