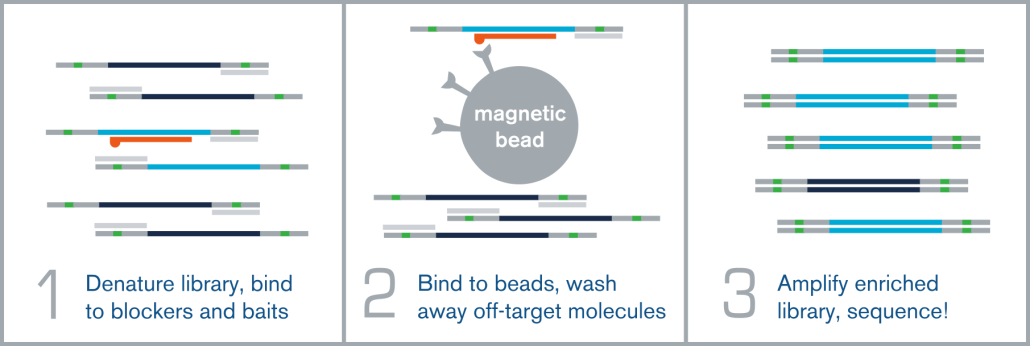
Hybridization capture is integrated into the overall next generation sequencing workflow immediately before sequencing on an NGS platform, such as Illumina. A fully sequenceable, barcoded/indexed NGS library (or pool of multiple libraries) is denatured, and allowed to anneal to complementary target-specific biotinylated probes/baits. These bait:library complexes are then bound to streptavidin-coated magnetic beads via the biotin on the probes, which are washed to remove non-specifically bound molecules. The remaining “enriched” library molecules are then released from the baits and amplified before sequencing.
Note! You may know the “hybridization capture” technique by another name, such as:
- Target enrichment
- Target capture
- Probe capture
- Exon capture
- Capture sequencing / sequence capture
- Hybridization sequencing / hyb-seq
- Hybridization capture / hyb-cap
 Linkedin
Linkedin Place order
Place order Quote request
Quote request Support
Support